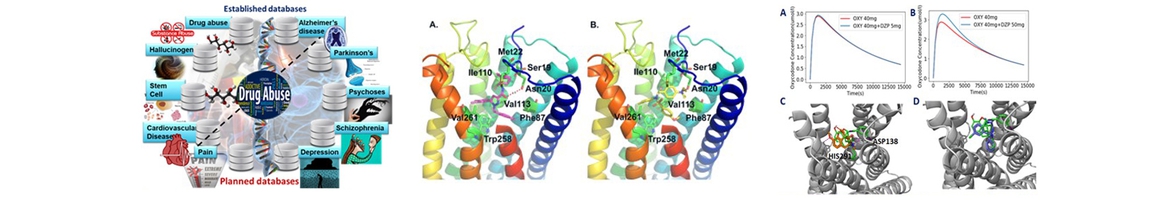
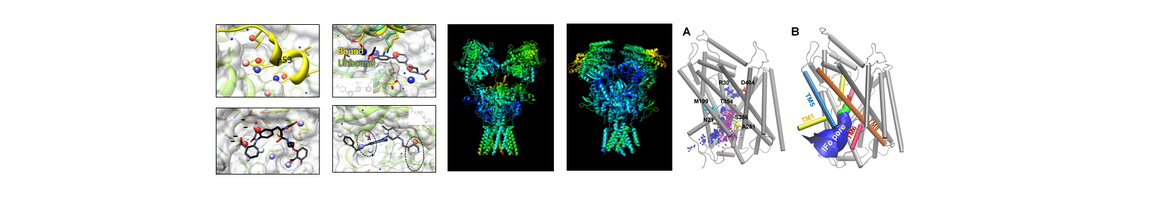
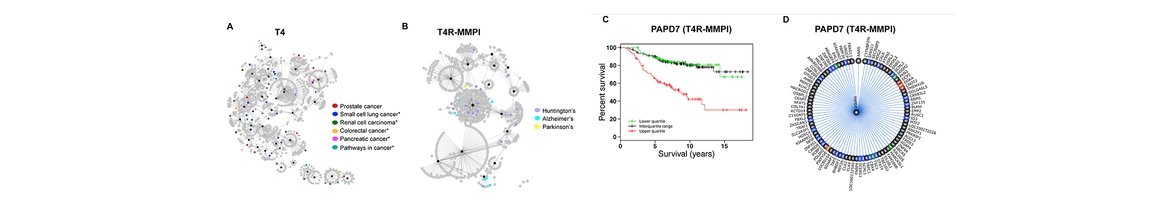
Structure, Function and Pharmacology of Neurotransmitter Reuptake Systems
Amara, Susan
U.S. National Institute of Mental Health
 Their laboratory has shown that amphetamines trigger the internalization of the dopamine transporter (DAT) by a series of intracellular events that are distinct from the generally established actions of amphetamines to inhibit dopamine (DA) uptake or to increase DA efflux. We have found that when applied to cell lines, cultured DA neurons or midbrain slices, amphetamine activates the small GTPases, RhoA and Rac-1 and triggers internalization of the dopamine transporter (DAT) by a specialized internalization pathway that requires the activation of the small GTPase, RhoA. Intriguingly, amphetamine must be transported into the cell to have these effects and its actions are actually blocked by cocaine, a drug that inhibits DAT and prevents amphetamine entry. We have also found that elevation of cAMP, via DA receptors or by amphetamine-induced adenylate cyclase activation, inactivates RhoA and limits carrier internalization, consistent with roles for PKA- and Rho-dependent signaling in mediating the actions of amphetamines in dopamine neurons. Our observations have also suggested the existence of a novel intracellular target for amphetamines and suggest new cellular pathways to target in order to disrupt amphetamine action. In recent studies we have established that a G-protein coupled trace amine receptor (TAAR1) serves as a direct intracellular target for amphetamines in dopamine neurons. Using transgenic mouse lines lacking the TAAR1 receptor we have shown that the intracellular effects of amphetamine, including both the elevation in cAMP and the increased RhoA activity, depend absolutely upon TAAR1 activation. We have shown that the when activated by amphetamine within the cell, TAAR1 couple through a G-protein alpha subunit, known as G13, and we have continued our efforts to identify the subcellular compartment where TAAR1 signaling takes place. In other studies within the group we have shown that G-protein beta-gamma subunits released when G-protein-coupled receptors are activated bind directly to the DAT and enhance dopamine efflux. Using cell permeable peptide fragments and mutagenesis of the DAT we have been able to define the transporter domains required for this interaction and to develop structural models for how this interaction may facilitate dopamine efflux by the transporter. Recent work has demonstrated that release of G-beta-gamma subunits triggered by endogenous receptor activation is sufficient to enhance DA efflux in cultured cells and neurons. We have also observed that the same amphetamine-activated RhoA-dependent mechanism downregulates a glutamate transporter, EAAT3, present on the surface of dopamine neurons. We have identified the EAAT3 peptide sequence responsible for this regulation, generated a cell-permeant fusion protein that blocks internalization and have used it to explore the effects of amphetamine on excitatory neurotransmission in brain slices. These studies have provided new tools to distinguish the effects of amphetamine on dopaminergic and glutamatergic signaling. We have also compared the effects of various amphetamine compounds on the activation of cellular signaling pathways. Comparison of the effects of methamphetamine on glutamate transport activity to those of amphetamine indicate that while both treatments lead to a loss of cell-surface EAAT3, the effects of methamphetamine are much broader and do not depend on the expression of the DAT. These findings provide an explanation for the broader, more devastating effects of methamphetamine: unlike amphetamine, methamphetamine has the capacity to alter EAAT3 surface expression and regulate excitatory neurotransmission, not only in dopamine neurons, but also in many other neuronal cell types within the brain. Glutamate transporters (also known as excitatory amino acid transporters or EAATs) present at the surface of neurons and supporting glial cells regulate the extracellular concentration of glutamate, the major excitatory neurotransmitter in the brain. By transporting glutamate back into the cell, these carrier proteins prevent glutamate from reaching toxic levels and also limit the extent and duration of transmitter signaling during glutamatergic neurotransmission. These carriers have an additional function in that they possess an anion channel activity that can regulate cellular excitability, which enables them to serve as sensors of glutamate levels outside the cell. Our laboratory has used site-directed mutagenesis, sulfhydryl modification, and chemical cross-linking approaches together with biochemical, and electrophysiological analyses of the mammalian carriers to examine the structural domains required for substrate transport and ion permeation. Recent work has been directed at understanding the mechanism and structural basis of anion channel activation. We initially identified a positively-charged arginine residue in transmembrane domain 7 as an essential element in the anion channel gating mechanism. Substitution of this residue with a negatively-charged amino acid, eliminates sodium- and substrate-dependent anion channel gating, and drives the channel into a substrate-independent constitutively open state. Using a computationally-derived homology model we identified a larger network of charged residues that are required to stabilize the closed state of the channel and, when examined experimentally, appear to be intrinsic elements of the channel gating mechanism. Overall, our data suggest that anion channel gating occurs through a transition from intermediate conformations that are closely linked to transport.
Their laboratory has shown that amphetamines trigger the internalization of the dopamine transporter (DAT) by a series of intracellular events that are distinct from the generally established actions of amphetamines to inhibit dopamine (DA) uptake or to increase DA efflux. We have found that when applied to cell lines, cultured DA neurons or midbrain slices, amphetamine activates the small GTPases, RhoA and Rac-1 and triggers internalization of the dopamine transporter (DAT) by a specialized internalization pathway that requires the activation of the small GTPase, RhoA. Intriguingly, amphetamine must be transported into the cell to have these effects and its actions are actually blocked by cocaine, a drug that inhibits DAT and prevents amphetamine entry. We have also found that elevation of cAMP, via DA receptors or by amphetamine-induced adenylate cyclase activation, inactivates RhoA and limits carrier internalization, consistent with roles for PKA- and Rho-dependent signaling in mediating the actions of amphetamines in dopamine neurons. Our observations have also suggested the existence of a novel intracellular target for amphetamines and suggest new cellular pathways to target in order to disrupt amphetamine action. In recent studies we have established that a G-protein coupled trace amine receptor (TAAR1) serves as a direct intracellular target for amphetamines in dopamine neurons. Using transgenic mouse lines lacking the TAAR1 receptor we have shown that the intracellular effects of amphetamine, including both the elevation in cAMP and the increased RhoA activity, depend absolutely upon TAAR1 activation. We have shown that the when activated by amphetamine within the cell, TAAR1 couple through a G-protein alpha subunit, known as G13, and we have continued our efforts to identify the subcellular compartment where TAAR1 signaling takes place. In other studies within the group we have shown that G-protein beta-gamma subunits released when G-protein-coupled receptors are activated bind directly to the DAT and enhance dopamine efflux. Using cell permeable peptide fragments and mutagenesis of the DAT we have been able to define the transporter domains required for this interaction and to develop structural models for how this interaction may facilitate dopamine efflux by the transporter. Recent work has demonstrated that release of G-beta-gamma subunits triggered by endogenous receptor activation is sufficient to enhance DA efflux in cultured cells and neurons. We have also observed that the same amphetamine-activated RhoA-dependent mechanism downregulates a glutamate transporter, EAAT3, present on the surface of dopamine neurons. We have identified the EAAT3 peptide sequence responsible for this regulation, generated a cell-permeant fusion protein that blocks internalization and have used it to explore the effects of amphetamine on excitatory neurotransmission in brain slices. These studies have provided new tools to distinguish the effects of amphetamine on dopaminergic and glutamatergic signaling. We have also compared the effects of various amphetamine compounds on the activation of cellular signaling pathways. Comparison of the effects of methamphetamine on glutamate transport activity to those of amphetamine indicate that while both treatments lead to a loss of cell-surface EAAT3, the effects of methamphetamine are much broader and do not depend on the expression of the DAT. These findings provide an explanation for the broader, more devastating effects of methamphetamine: unlike amphetamine, methamphetamine has the capacity to alter EAAT3 surface expression and regulate excitatory neurotransmission, not only in dopamine neurons, but also in many other neuronal cell types within the brain. Glutamate transporters (also known as excitatory amino acid transporters or EAATs) present at the surface of neurons and supporting glial cells regulate the extracellular concentration of glutamate, the major excitatory neurotransmitter in the brain. By transporting glutamate back into the cell, these carrier proteins prevent glutamate from reaching toxic levels and also limit the extent and duration of transmitter signaling during glutamatergic neurotransmission. These carriers have an additional function in that they possess an anion channel activity that can regulate cellular excitability, which enables them to serve as sensors of glutamate levels outside the cell. Our laboratory has used site-directed mutagenesis, sulfhydryl modification, and chemical cross-linking approaches together with biochemical, and electrophysiological analyses of the mammalian carriers to examine the structural domains required for substrate transport and ion permeation. Recent work has been directed at understanding the mechanism and structural basis of anion channel activation. We initially identified a positively-charged arginine residue in transmembrane domain 7 as an essential element in the anion channel gating mechanism. Substitution of this residue with a negatively-charged amino acid, eliminates sodium- and substrate-dependent anion channel gating, and drives the channel into a substrate-independent constitutively open state. Using a computationally-derived homology model we identified a larger network of charged residues that are required to stabilize the closed state of the channel and, when examined experimentally, appear to be intrinsic elements of the channel gating mechanism. Overall, our data suggest that anion channel gating occurs through a transition from intermediate conformations that are closely linked to transport.